Announcing the Allele Frequency Aggregator (ALFA) Project as part of the Bio-IT World 2022 Hackathon: Visualization of NCBI ALFAVariants
Join NCBI at the Bio-IT World 2022 Hackathon on May 4-5, 2022 to learn about and work with data from our ALFA project! The primary goal of this hackathon project is to develop a novel tool, app, or approach to explore and visualize NCBI ALFA variants and allele frequency for 12 different human populations. We aspire to create a new helpful variant interpretation resource for the clinical and research communities.
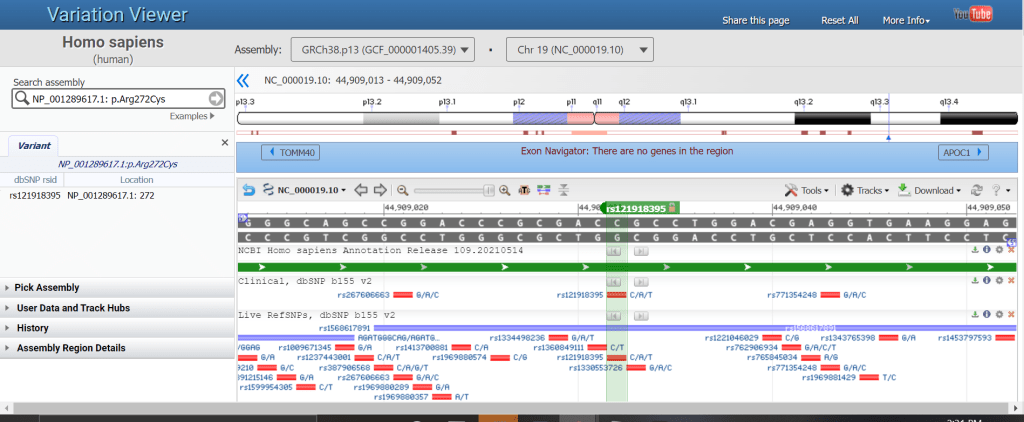
If you’ve ever tried searching for a genomic location in NCBI’s Genome Data Viewer (GDV) or Variation Viewer and found that your search term didn’t work, it’s time to try again! We recently expanded support for searches in our genome browsers using non-NCBI identifiers such as HGVS patterns (e.g. NM_001318787.2:c.2258G>A) and Ensembl IDs. You can also search by chromosome coordinates, cytogenetic band, assembly scaffold/component, disease/phenotype, dbSNP identifier, or RefSeq transcript/protein accession. We’ve gathered example searches in the table below.
When you search by single coordinate, SNP or dbVar ID, or HGVS, the browser view zooms to the location of the search result. A marker is automatically created to identify the searched position. For HGVS, the marker is labelled with the corresponding rsID, if there is one.
Figure 1. Variation Viewer showing results of search by an HGVS pattern, NP_001289617.1: p.Arg272Cys.
As always, please contact us if you have additional questions or suggestions about this or any other feature in GDV or Variation Viewer. You can use the Feedback button on the page or write to the NCBI Help Desk directly.
Two up-and-coming NCBI resources will be featured in videos, surveys and live events at the American Society for Human Genetics (ASHG) 2020 Annual Meeting. Come and watch on-demand videos in the CoLab Theater. Then, let us know what you think and how you do or might use these resources by either taking an online survey or joining us for the CoLab Live! Events on Thursday, October 29, 2020.
dbVar, NCBI’s database of large-scale genetic variants, has a new track hub for viewing and downloading structural variation (SV) data in popular genome browsers. Initial tracks include Clinical and Common SV datasets. dbVar’s new track hub can be viewed using NCBI’s Genome Data Viewer through the “User Data and Track Hubs” feature (Figure 1) and other genome browsers by selecting “dbVar Hub” from the list of public tracks or by specifying the following URL.
Figure 1. Loading the dbVar track hub in the Genome Data Viewer. The Track Hubs feature on the left-hand column of the browser allow you to add the track by searching for it or by entering the direct URL. You can select the specific tracks — for example, “NCBI curated common SVs: All populations” — to load from the Configure Track Hubs dialog.Continue reading “dbVar clinical and common structural variants track hub now available”→
The latest dbVar data release includes the Genome in a Bottle benchmark structural variant (SV) callset (pre-print Zook et al. 2019) – a highly scrutinized, carefully curated set of 12,745 sequence-resolved deletions, insertions, and delins variants from Personal Genome Project Ashkenazi trio son HG002. The data serve as a robust benchmark standard with which to measure the performance of sequencing and variant-calling pipelines. It “reliably identifies both false negatives and false positives in high-quality SV callsets” (pre-print Zook et al. 2019) that are based on short-, linked-, and long-read sequencing as well as optical mapping.
We’ve expanded the catalog of clinically relevant structural variants (SV) in dbVar by adding 57,520 ClinVar records. You can access the newly added data through study nstd102.
The updated collection includes:
20,000 new SVs, and more than 37,000 copy number variants (CNV) observed in ClinGen laboratories during routine cytogenomic laboratory testing that were previously accessioned separately at dbVar
15,000 SVs asserted as ‘Pathogenic’ or ‘Likely pathogenic’ for thousands of clinical genetic disorders including breast, ovarian, and colon cancers; hypercholesterolemia; schizophrenia; Duchenne Muscular Dystrophy; autism spectrum disorders; and many others
links to more than 1,600 related PubMed articles and thousands of related data records in ClinVar, OMIM, GeneReviews, MedGen, MeSH, etc.
You can browse dbVar studies on the web or download the data. We provide dbVar data in a number of standard formats (VCF, GVF, and TSV) mapped to assemblies GRCh38, GRCh37, and NCBI36 allowing you perform analysis using standard tools and integrate the data into your bioinformatic workflows.
Visit our Walkthrough pageto learn how to use these new dbVar data to help interpret structural variation in your favorite gene or genomic region.
Would you like to compare and analyze your data with known structural variants (SV) in NCBI’s database of genomic structural variation (dbVar)? Now there are easy-to-use files containing non-redundant (NR) deletions, duplications, and insertions aggregated from across studies in dbVar. The files are available for human assembly versions GRCh37 and GRCh38. Descriptions of the NR data are available on GitHub.
The NR files are available for FTP download in BED, BEDPE, and custom tab-separated formats, designed to be compatible with many popular tools and browsers. To help users get started, we have developed tutorials for UCSC Genomic Browser, Galaxy web-based analysis platform, NCBI Sequence Viewer, and command-line BEDtools.
An upcoming release will include annotations including genes, regulatory regions, and more. Have a favorite annotation you’d like to see? Send us your suggestions by contacting dbVar directly or open a GitHub issue. We also welcome comments and other improvement suggestions.
dbVar non-redundant SV (NR SV) datasets include more than 2.2 million deletions, 1.1 million insertions, and 300,000 duplications. These data are aggregated from over 150 studies including 1000 Genomes Phase 3, Simons Genome Diversity Project, ClinGen, ExAC, and others. You can use NR SV data files to filter and annotate variants in a broad range of applications:
Clinicians can easily filter patients’ genome data to find SV that overlap with variants previously reported as clinically significant.
Researchers can compare the results of their own genome-wide SV surveys with dbVar NR data to identify variants that are novel or rare, those which may be pathogenic, and in some cases obtain allele frequencies for matching variants. Users can also annotate SV data with NR SV and other genomic annotations to prioritize those variants most likely to impact biological function.
Developers of variant analysis pipelines can use dbVar NR data to help identify novel variants, calibrate their algorithms, or simply integrate the data into downstream analysis tools and workflows.
dbVar’s NR SV reference data are updated monthly. These updates include new database submissions. We welcome your feedback on the content and usability of these files so that we can improve them.
For more information, please see our GitHub site, which includes brief tutorials and access to NR SV datasets by >FTP.
The new services are faster, better at handling variants in repeat regions, and scalable to accommodate the continued explosive growth of variation volume. You can find more information about the services in the initial blog post and online SPDI document.
If you would like to report any issues related to these new services and/or would like to provide comments, please write to [email protected].
If you have any specific questions about the NCBI site in general, contact us at [email protected].
We appreciate your continued support and interaction with the NCBI tools.
dbVar has generated known structural variants (SV) datasets for use in comparisons with user data to aid variant calling, analysis and interpretation.
Files containing Non-Redundant (NR) deletions, insertions, and duplications are now available on GitHub. Additional separate files include preliminary annotations of overlap with ACMG59 genes. All files are in tab-delimited text format.
We encourage you to test these files and provide feedback, either on GitHub or by email.