Version 1.1.0 "Parsley" Update
Description
This is version 1.1.0 Parsley.
This release contains results of valence parameter re-fitting, with small modifications in the input force field. result.offxml in Assets is the resulting force field file,version 1.1.0 Parsley from the re-fitting. The modifications include the following;
(1) Addition of three new bond and angle terms, a22a, b14a and b36a;
a22a, b14a and b36a were added to effectively consider conjugation effect.
Separating a22a from a22 was intoduced to effectively describe conjugation effect of N=C=S, which makes the equilibrium angles of a22a relatively larger than a22.
<Angle smirks="[*:1]~[#7X2+0:2]~[*:3]" angle="120.0 * degree" k="140.0 * mole**-1 * radian**-2 * kilocalorie" id="a22" parameterize="k,angle"/>
<Angle smirks="[*:1]~[#7X2+0:2]~[#6X2:3](~[#16X1])" angle="120.0 * degree" k="140.0 * mole**-1 * radian**-2 * kilocalorie" id="a22a" parameterize="k,angle"/>
Separating b14a from b14 was intoduced to effectively describe resonance of single bonds between sp2 C and O with negative 1 charge.
<Bond smirks="[#6:1]-[#8:2]" length="1.41 * angstrom" k="640.0 * angstrom**-2 * mole**-1 * kilocalorie" id="b14" parameterize="k,length"/>
<Bond smirks="[#6X3:1]-[#8X1-1:2]" length="1.41 * angstrom" k="640.0 * angstrom**-2 * mole**-1 * kilocalorie" id="b14a" parameterize="k,length"/>
Separating b36a from b36 was intoduced to effectively describe resonance of double bonds between N with positive 1 charge and N with negative 1 charge.
<Bond smirks="[#7:1]=[#7:2]" length="1.3 * angstrom" k="680.0 * angstrom**-2 * mole**-1 * kilocalorie" id="b36" parameterize="k,length"/>
<Bond smirks="[#7+1:1]=[#7-1:2]" length="1.27 * angstrom" k="760.0 * angstrom**-2 * mole**-1 * kilocalorie" id="b36a" parameterize="k,length"/>
(2) Modification of periodicity for N-N rotations;
Periodicity of t128 has been changed to be represented by twofold and threefold periodicity, while t129, t130 and t131 has been changed to have twofold periodicity.
<Proper smirks="[*:1]~[#7X2:2]-[#7X3:3]~[*:4]" periodicity1="3" periodicity2="2" phase1="0.0 * degree" phase2="180.0 * degree" k1="0.625 * mole**-1 * kilocalorie" k2="0.0 * mole**-1 * kilocalorie" id="t128" idivf1="1.0" idivf2="1.0" parameterize="k1,k2"/>
<Proper smirks="[*:1]=[#7X2:2]-[#7X2:3]=[*:4]" periodicity1="2" phase1="180.0 * degree" k1="1.4 * mole**-1 * kilocalorie" id="t129" idivf1="1.0" parameterize="k1"/>
<Proper smirks="[*:1]~[#7X2:2]=,:[#7X2:3]~[*:4]" periodicity1="2" phase1="180.0 * degree" k1="6.0 * mole**-1 * kilocalorie" id="t130" idivf1="1.0" parameterize="k1"/>
<Proper smirks="[*:1]~[#7X3+1:2]=,:[#7X2:3]~[*:4]" periodicity1="2" phase1="180.0 * degree" k1="3.0 * mole**-1 * kilocalorie" id="t131" idivf1="1.0" parameterize="k1"/>
(3) Addition of missing improper torsions (i2a, i3a, i3b, i5a) with the associated torsions (t51a,t51b, t51c).
New torsion t51a was added to describe sp3 C connected to trivalent N which is connected to N=C and t51b was added for sp3 C connected to trivalent N which is connected to N=N or N=O. For sp3 C and trivalent N bond connected to 5-membered hetero-aromatic ring, the torsion was separately assigned to t51c.
<Proper smirks="[*:1]-[#6X4:2]-[#7X3:3]-[*:4]" periodicity1="3" phase1="0.0 * degree" k1="0.3 * mole**-1 * kilocalorie" id="t51" idivf1="1.0" parameterize="k1"/>
<Proper smirks="[*:1]-[#6X4:2]-[#7X3:3]-[#7X2:4]=[#6]" periodicity1="3" periodicity2="2" phase1="0.0 * degree" phase2="180.0 * degree" k1="0.3 * mole**-1 * kilocalorie" k2="0.0 * mole**-1 * kilocalorie" id="t51a" idivf1="1.0" idivf2="1.0" parameterize="k1,k2"/>
<Proper smirks="[#1:1]-[#6X4:2]-[#7X3:3]-[#7X2:4]=[#6]" periodicity1="3" periodicity2="2" phase1="0.0 * degree" phase2="180.0 * degree" k1="0.3 * mole**-1 * kilocalorie" k2="0.0 * mole**-1 * kilocalorie" id="t51ah" idivf1="1.0" idivf2="1.0" parameterize="k1,k2"/>
<Proper smirks="[*:1]-[#6X4:2]-[#7X3:3]-[#7X2:4]=[#7X2,#8X1]" periodicity1="3" periodicity2="2" phase1="0.0 * degree" phase2="180.0 * degree" k1="0.3 * mole**-1 * kilocalorie" k2="0.0 * mole**-1 * kilocalorie" id="t51b" idivf1="1.0" idivf2="1.0" parameterize="k1,k2"/>
<Proper smirks="[#1:1]-[#6X4:2]-[#7X3:3]-[#7X2:4]=[#7X2,#8X1]" periodicity1="3" periodicity2="2" phase1="0.0 * degree" phase2="180.0 * degree" k1="0.3 * mole**-1 * kilocalorie" k2="0.0 * mole**-1 * kilocalorie" id="t51bh" idivf1="1.0" idivf2="1.0" parameterize="k1,k2"/>
<Proper smirks="[*:1]-[#6X4:2]-[#7X3$(*@1-[*]=,:[*][*]=,:[*]@1):3]-[*:4]" periodicity1="2" phase1="180.0 * degree" k1="2.0 * mole**-1 * kilocalorie" id="t51c" idivf1="1.0" parameterize="k1"/>
<Proper smirks="[#1:1]-[#6X4:2]-[#7X3$(*@1-[*]=,:[*][*]=,:[*]@1):3]-[*:4]" periodicity1="2" phase1="180.0 * degree" k1="2.0 * mole**-1 * kilocalorie" id="t51ch" idivf1="1.0" parameterize="k1"/>
There was an addition of new improper torsions as well. i2a was newly added to describe trivalent N centers connected to pi-bond forming S or P. i3a was added for trivalent N centers connected to a pi-bond-forming N and i3b was separately defined for trivalent N centers inside 5-membered hetero-aromatic ring.
<Improper smirks="[*:1]~[#7X3$(*~[#15,#16](!-[*])):2](~[*:3])~[*:4]" periodicity1="2" phase1="180.0 * degree" k1="1.1 * mole**-1 * kilocalorie" id="i2a" />
<Improper smirks="[*:1]~[#7X3$(*~[#7X2]):2](~[*:3])~[*:4]" periodicity1="2" phase1="180.0 * degree" k1="1.1 * mole**-1 * kilocalorie" id="i3a" />
<Improper smirks="[*:1]~[#7X3$(*@1-[*]=,:[*][*]=,:[*]@1):2](~[*:3])~[*:4]" periodicity1="2" phase1="180.0 * degree" k1="10.5 * mole**-1 * kilocalorie" id="i3b" />
(4) t19 removal of redundant periodicity.
A redundant threefold periodicity component of t19 has been removed.
<Proper smirks="[#1:1]-[#6X4:2]-[#6X3:3]=[#6X3:4]" periodicity1="3" periodicity2="1" phase1="180.0 * degree" phase2="0.0 * degree" k1="0.38 * mole**-1 * kilocalorie" k2="1.15 * mole**-1 * kilocalorie" id="t19" idivf1="1.0" idivf2="1.0" parameterize="k1,k2"/>
Fitting Data
- Roche set: 686 1-D torsion profiles; 936 optimized geometries; 669 vibrational frequencies
- Coverage set: 412 1-D torsion profiles; 842 optimized geometries; 265 vibrational frequencies
- outside of the two sets: 19 1-D torsion profiles
Results
The objective function decreased from 2.63784e+04 to 4.63632e+03 in 18 steps.
1. Fitting Results
1-1. Addition of three new bond and angle terms, a22a, b14a and b36a
The plots below shows the result of addition of the subgroup a22a .The optimized equilibrium angle values for a22 and a22a are quite different (118 and 143 degree respectively) which validates the sub-grouping.

Same results were observed in b14 and b36 cases. The final equilibrium bond values are quite different between supersets and their subsets. (Bad initial guesses on b14a and b36a are suspected to be the major factor of slightly off fitting results.)
1-2. Modification of periodicity for N-N rotations
While inspecting the benchmark result of Parsley, significantly large RMSD of MM optimized geometry from QM optimized geometry was found in the molecule Cc1ccc(cc1)N/N=N/c2ccccn2 and the main contribution of the high RMSD value came from dihedral angles. t130 (SMIRKS: [*:1]~[#7X2:2]=,:[#7X2:3]~[*:4]) assigned to 10-13-14-15 torsion was found to have its periodicity 1 and the issue was resolved by simply modifying the periodicity to 2. Also it was confirmed that the modification of the periodicity led to a slight decrease in the final objective value in valence parameter fitting.

1-3. Addition of missing improper torsions (i2a, i3a, i3b, i5a) with the associated torsions (t51a,t51b, t51c)
We found that Parsley didn't have an improper torsion for the trivalent nitrogen center in tetrazole. But simply Adding the missing improper terms (trivalent nitrogen centers connected to pi-bond-forming atom) couldn't make the nitrogen center planar even with unphysically large k value due to the contribution from torsions with inappropriate periodicity. The torsions assigned to the bond between the trivalent nitrogen and its attached sp3 carbon were t51, whose periodicity is set to 3. Threefold periodicity is reasonable when both ends of central bond are threefold axis. However, when the nitrogen in the center bond is conjugated, forming the nitrogen end twofold axis, like the trivalent nitrogen in tetrazole, threefold periodicity is not able to describe the planar nitrogen center successfully. In other words, the t51 term in the previous force field needed to be seperated into subgroups.
Note: since 5-membered hetero aromaticity is pretty common and one of the important functional groups in drug molecules, we separately defined proper and improper torsions for the trivalent nitrogens inside 5-membered heteroaromatics.

2. Benchmark Results
2-1. Benchmark Primary Set
Objective value of single point calculation on Primary set for v1.1.0 is 936.17, which is lower than v1.0.0 (948.01) and much lower than initial force field (1434.54)
2-1-1. Optgeo Targets
To investigate the improved performance of the new parameter set in reproducing QM optimized geometries over Parsley, the weighted root-mean-square error (WRMSE) of each molecule, which is weighted root-mean-square deviation between internal coordinates of MM optimized geometry and QM optimized geometry was calculated for each parameter set then comparison of the WRMSE values was carried out.
( Metrics for bond, angle, improper torsion are set to be 0.05 Angstrom, 8 degree and 20 degree respectively and torsion contributions were intentionally excluded.)
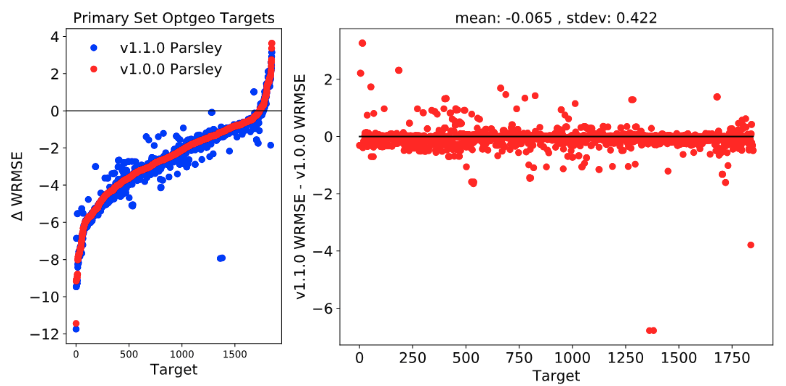
Note: y values in the plots (Δ weighted RMSE) are the changes in th weighted RMSE from initial force field to optimized force field; Negative y value indicates better reproduction in the optimized force field compared to the initial ff.
There has been a slight improvement on reprodicing QM optimized geometries.
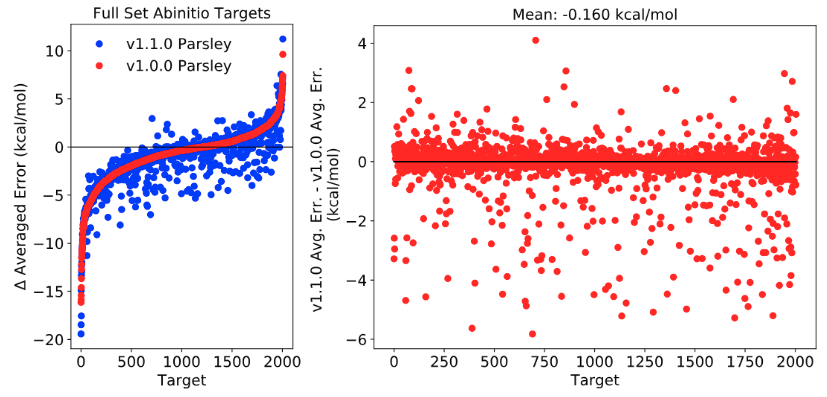
2-1-2. Abinitio Targets
To investigate the improved performance of the new parameter set in reproducing QM relative energies between conformers over Parsley, absolute QM vs MM relative energies differences averaged over conformers was calculated.

Note: y values in the plots (Δ Average Error) are the changes in the average error from initial force field to optimized force field; Negative y value indicates better reproduction in the optimized force field compared to initial ff.
There has been no improvement on reprodicing QM relative energies of conformers.
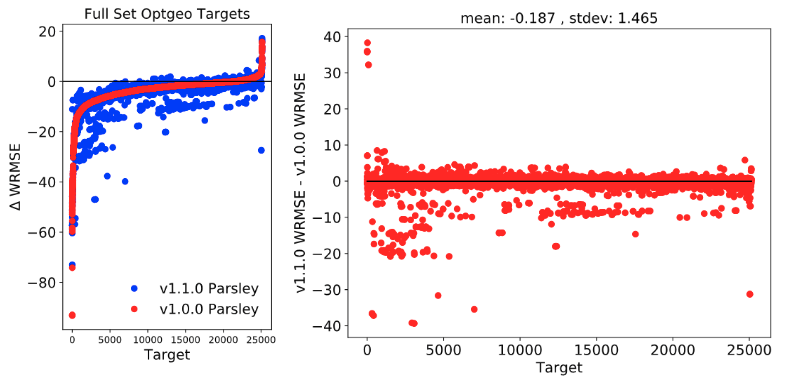
2-2. Benchmark Full Set
Objective value of single point calculation on Primary set for v1.1.0 is 20096.94, which is lower than v1.0.0 (20672.00) and much lower than initial force field (29471.05)
2-2-1. Optgeo Targets
There has been a slight improvement on reprodicing QM optimized geometries.
2-2-2. Abinitio Targets
There has been a slight improvement on reprodicing QM relative energies of conformers.